Hello !
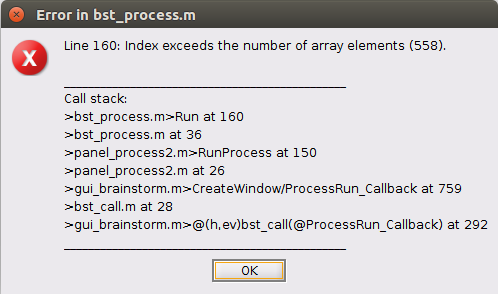
When I put the baseline files (A) and compare it to B tab in the process 2 tab. Weirdly the process of baseline normalization in standardization is "blocked" or just not accessible. Somehow, if I click on it it still works but at the end I get this error message and the normalized files dont appear until a loooong time ...
I have to do the same with EMG files and everything is going smoothly...
Hi Beatrice,
This process requires the same amount of input files in process box A and B. This is probably why you have it greyed out. If you want to use one file and compare it to many others, make sure to duplicate the file to balance your 2 process boxes. If that doesn't make sense or you think your error is not related to this, please post a screenshot of your process box to give us an idea of what you're trying to do.
I hope this helps,
Martin
Hi Martin,
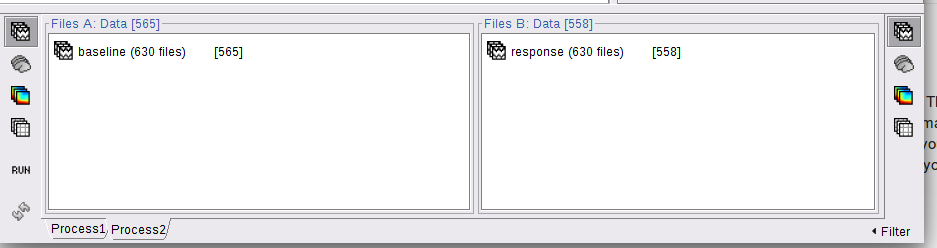
Here's the screenshot, same amount of files loaded but different amount in the brackets...
This is because some of the epochs were marked as bad and are therefore excluded from the file selection:
https://neuroimage.usc.edu/brainstorm/Tutorials/Epoching#Import_in_database
The problem is that the list of bad trials is not the same in your baselines and your responses. I'm not sure what's the best approach for going around this problem.
- You could manually mark as bad or delete all the responses+baselines for which there is either the baseline or the response that is bad.
- You could mark all the trials as good (but you would lose the benefits of the preprocessing you've done before)
- A more technical option would consist in editing the file structure that contains the list of bad trials (the files brainstormstudy.mat), run your baseline correction, and finally restore the list of bad trials. If you are confident with editing directly the files structures with Matlab, I could give you more directions if you need.
Note that when processing MEG/EEG recordings, the process "baseline normalization" is useful only with the option "DC offset correction". The other types of normalizations are indicated for source results or time-frequency maps.
Baseline is computed from exactly the same events (stim) but epoch time for baseline set as -199.2- 0 ms and response -199.2-199.2. That could explain why some more bad segments are included in the response files.. no ?
Maybe this option is not so bad as there's around 7-6 different files between the two. I just delete directly form the list ?
How come the process still seem to work with time, i come back and the response | zscore appears..
Baseline is computed from exactly the same events (stim) but epoch time for baseline set as -199.2- 0 ms and response -199.2-199.2.
Then do not import the baseline as separate epochs. If all your "baseline" epochs are only duplicates of the recordings already included in the "response" epochs, then they are useless. Proceed as described in the introduction tutorials: https://neuroimage.usc.edu/brainstorm/Tutorials/Epoching
Then select all your epochs in Process1 and select the process "Pre-process > Remove DC offset" with a baseline [-199, 0].