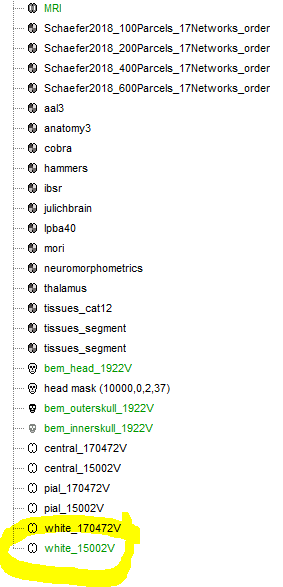
I need to load the currently selected surface file for each subject in my database from "brainstormsubject.mat" and I have noticed that if after the initial segmentation with CAT12 the selected file is "tess_cortex_white_low" (see figure) with no user input this information will not be saved in "brainstormsubject.mat" and it will be selected on loading the database by default (I guess by being the last available surface file)
There is something that is not working as expected in your example.
After importing the CAT12 files, the function import_anatomy_cat_2020 explicitly sets the default cortex surface to be the low resolution CENTRAL surface, not the white one.
Please make sure that your version of Brainstorm is up-to-date (from a few days ago). Make sure CAT and SPM are also up-to-date.
If the white file is repeatedly selected at the end of the CAT12 segmentation, there is problem somewhere, that we need to fix. But first we need to be able to reproduce it on our end.
Put a breakpoint at line 552 in import_anatomy_cat_2020.m (click on the line number), run the segmentation, when the debugger stops at the end of the CAT12 execution, execute the code line by line to make sure it really sets the default surface to be the central one.
Hi, that's very strange, but I wasn't able to reproduce this with the subjects in which this happened. I made a new subject with the same MRI and this time it all behaved as expected... maybe it's because I did it through the GUI this time? Usually I use this code to automate it:
for nSubject = 1:length(existing_subjects)-1
% Load subject to get info
indexN = find(contains(names,existing_subjects(nSubject).name));
selectedEEG = ids{indexN};
selectedGroup = groups{indexN};
sFiles = [existing_subjects(nSubject).name '\' selectedGroup '\data_temp_0001.mat'];
% If segmentation has already been done, skip
if isfile([folder_anat existing_subjects(nSubject).name '\tess_cortex_central_low.mat'])
continue
end
% Process: Segment MRI with CAT12
sFiles = bst_process('CallProcess', 'process_segment_cat12', sFiles, [], ...
'subjectname', existing_subjects(nSubject).name, ...
'nvertices', 15000, ...
'tpmnii', {'', 'Nifti1'}, ...
'sphreg', 1, ...
'vol', 1, ...
'extramaps', 0, ...
'cerebellum', 0);
% Save and display report
ReportFile = bst_report('Save', sFiles);
bst_report('Open', ReportFile);
end
In your script, sFiles should be a cell-array of strings rather than a string. Just enclose its definition within {}.
Other than this minor detail, I don't think there is any problem with your script.
The code executed from the GUI or the pipeline editor is the same.
I also noticed that from time to time, it was not the correct cortex surface that was selected at the end of the process, but I could never find a way to reproduce it.
Please let me know if you find a procedure to reproduce this incorrect selection (white surface instead of central).