I currently analyse the long latency auditory evoked potentials (LLAEPs) recorded with a white noise stimulus. I would like to check responses from P1, N1, P2 complex waves!
I was wondering if it was necessary (or optional) to apply a ‘‘Sinusoid removal’’ in my process before identifying the artifacts (heart, eyes) and importing raw files in database?
I had a problem in a new subject analysis. As usual, I have imported the data and reviewed the raw file. When I tired to detect the artifacts this message occurred in the command windows:
CTF> 272 MEG coil definitions set up.
CTF> WARNING: \rhythm.brams.umontreal.ca\labs\lab_hebert\Projet_Labo_Hébert\Étude Bouchons-Générateurs\Results MEG\PB006-pre_GAIN-shebert_20131119_01_AUX.ds\PB006-pre_GAIN-shebert_20131119_01_AUX.meg4 does not contain an integer number of trials
BST> Not enough digitized head points to perform automatic registration.
** Error: Line 61: view_surface_data (line 61)
** File not found in database: “”
**
** Call stack:
** >view_surface_data.m at 61
** >bst_figures.m>ReloadFigures at 1501
** >bst_call.m at 28
** >macro_methodcall.m at 39
** >bst_figures.m at 59
** >panel_record.m>ReloadRecordings at 994
** >panel_record.m>CallProcessOnRaw at 2241
** >panel_record.m>@(h,ev)CallProcessOnRaw(‘process_evt_detect_ecg’) at 165
**
I don’t know what’s happened because I did the same procedure: I used the default anatomy without MRI data! About the digitized head points, I normally add my polhemus after reviewed the raw file, detected artifacts and computed SSP (CTF channels > Digitized head points > Add points > headshape.eeg). Could you explain that?
I’ve just fixed this small bug. You can update Brainstorm (Help > Update).
It was nothing related with the processing, it happened when trying to refresh the display just because you had a figure with some surfaces opened.
In general, I would recommend importing the head points immediately after linking the continuous file to the database to avoid forgetting them.
Also, if you add manually the head shape, you have to run the registration based on the head shape manually (right-click on channel file > MRI registration > Refine using head points)
Considering your last post, I was wondering if it’s necessary to finish the registration of the head shape by the following process: (right-click on channel file > Digitized head points > Deform default anatomy to fit points). If so, do I need to select ‘‘wrap’’ or ‘‘scale’’?
If you don’t have the individual MRI of your subjects, it is indeed a good thing to deform the Colin27 template to match the shape of the subject’s head.
If you do this, I recommend you do NOT run the automatic registration first (Refine using head points) because it would most likely not perform well.
This automatic registration algorithm searches for an optimal fit between the headshape from the Polhemus and the one extracted from the MRI (head surface).
The algorithm is quite robust when you have the individual subject MR. But if the two surfaces are not representing the same person, the output can be completely random.
The recommended procedure in this case is:
Review raw file
Digitized head points > Add points
Right-click on channel file > MRI registration > Edit > do your best to align manually the green points and the grey head surface
Digitized head points > Deform default anatomy to fit points > Warp
The “Scale” menu should be used only if you don’t have enough head points, or if the output you get using “Warp” is really bad.
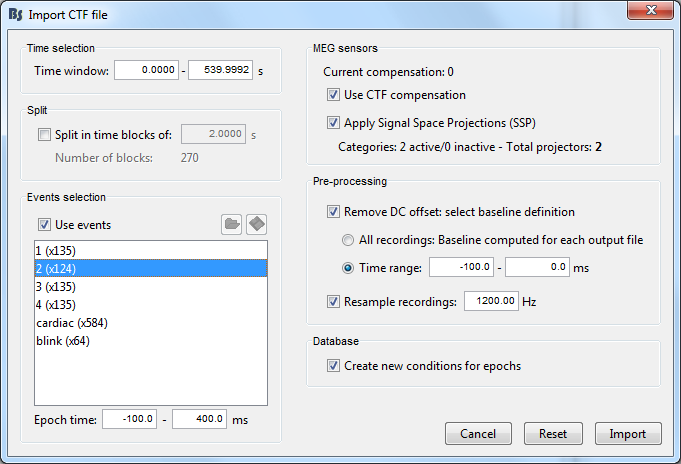
Considering I analyse LLAEPs, which best sampling should I use when I import my CTF file? By default, the resample recording is at 1200 Hz in the pre-processing window.
If you don’t need absolutely need to resample your recordings, I recommend you uncheck this resample option and leave the recordings at the original sampling rate.
The resampling operation alters the frequency contents of the recordings, it’s better to avoid it when possible.
When I import another Raw file for a same subject, do I need to add my polhemus points again?
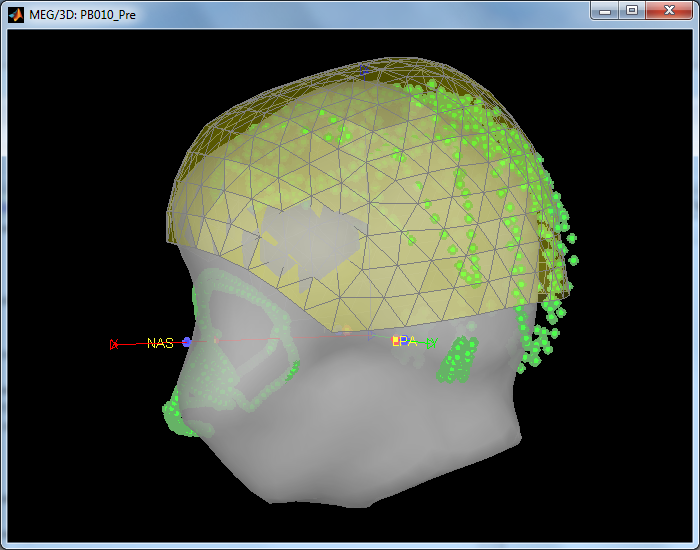
By Right-click on channel file for this raw file > MRI registration > Check, I can observe the deform default anatomy previously saved but my points don't fit with the shape!
Here a picture... What I suppose to do? keep the deform default anatomy or try to to align once again the green points and this grey head surface?
Run the automatic registration (MRI registration -> refine using headpoints)
Note that we don’t recommend using the “continuous mode” of the digitizing device.
You can find an example of head shape that works well with the Brainstorm algorithms on the Digitizer tutorial page: http://neuroimage.usc.edu/brainstorm/Tutorials/TutDigitize
I would like to have some advices for managing my subjects data in the protocol I've previously created.
Here the problem:
In a same protocol study, I've imported 6 subjects. They are split in 2 different groups (3 subj in each: Bouch vs Gene) depending of the therapy applied!
I've recorded the LLAEPs for each subject before and after the therapy. LLAEPs were recorded under 4 conditions (events 1, 2, 3 and 4), that is the sound level of the white noise (60, 70, 80, 90 dB SPL).
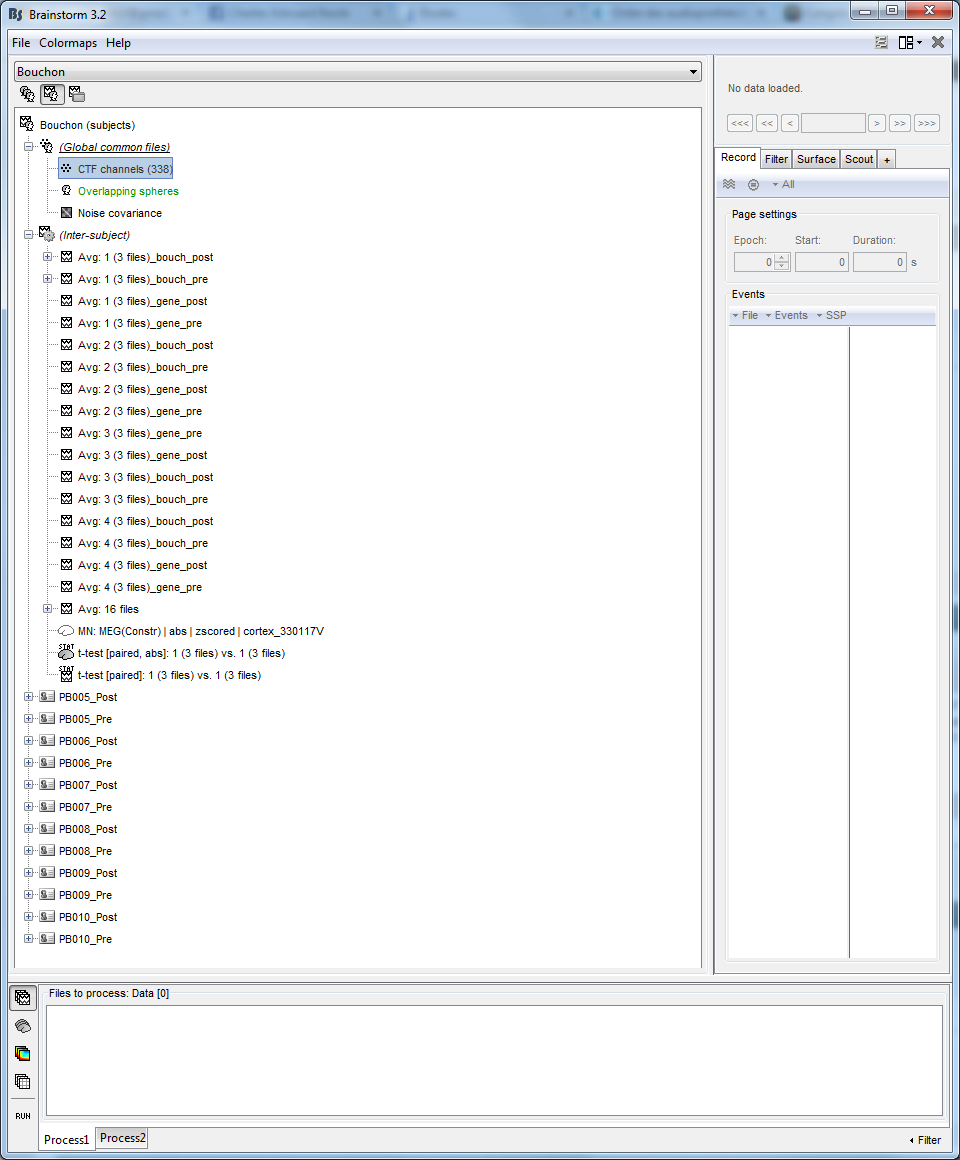
I would like to compare waves latencies and amplitudes for each group for each condition between pre and post session but in the protocol window, all my subjects were included in the same inter-subject link and there is a problem because when I compute sources it's not the same head model and noise covariance... So What is the best way to perform my comparison?
Here a picture:
You should not average MEG recordings at the sensor level as you do with EEG. The head is moving in a fixed array of sensors, therefore the same sensor is most likely not capturing the information from the same brain regions in two different runs. The proper way is to calculate your group averages at the source level. Select the same files in the Process1 tab, but select the “process sources” button on the left side of the panel.
This requires a first step of anatomy normalization (skip this if all your subjects are warped versions of the Colin27 brain): http://neuroimage.usc.edu/brainstorm/Tutorials/CoregisterSubjects
Then, for your database organization, the default “inter-subject” node is very limited. It is used to store all the files generated by the Brainstorm processes from multiple subjects. But after calculating your averages, you can move them from (inter-subject) to somewhere else that makes more sense to you.
For instance: create two additional subjects for storing the averages of the subject groups, with your four conditions each.
To move the files: 1) CTRL+X/CTRL+V or 2) drag and drop or 3) right-click > File > Cut/Paste
When you calculate your averages at the source level, you don’t have the question of what is the “common” channel file / head model / source model.
I was wondering the best way to average my sources:
put my subject sources in the process1 before or after apply a z-score standardization?
average my files by condition (grand average) or by trial group (grand average)?
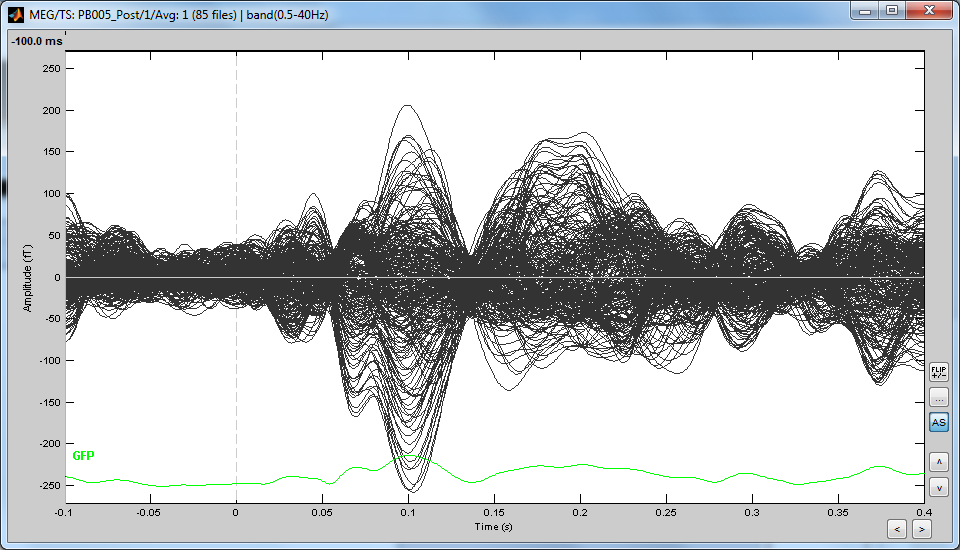
Also, I have another question. I would like to have an overview of my channels recording, and I was wondering if we could average all the channels in only one response? (see attached picture)
Both are acceptable. Mathematically, “average(z-score)” is a questionable but many people do it to get more comparable values between subjects.
You can try what you get with both option and see if it looks like it balancing more the input from the difference subjects…
This depends more on your database structure:
if you have one experimental condition per run OR only one run: by condition
if you have multiple experimental conditions AND multiple runs: by trial group
I've just edited my last message but I don't know if you saw it: I would like to have an overview of my channels recording, and I was wondering if we could average all the channels in only one response? (see attached picture)
Yes you can, but just to get a first overview of your recordings.
Keep in mind this is not an accurate way of averaging MEG recordings across subjects.
Francois